构建常用 index
构建索引是基因组比对中的一个关键步骤,它允许比对软件高效地查询和比对测序数据。不同的比对软件通常需要不同格式的索引,下面是几种常见比对软件的索引构建的流程。
snakemake 流程
运行 常见软件的索引将生成到参考基因所在目录中,类似 hisat2_index/ 的目录下。
流程图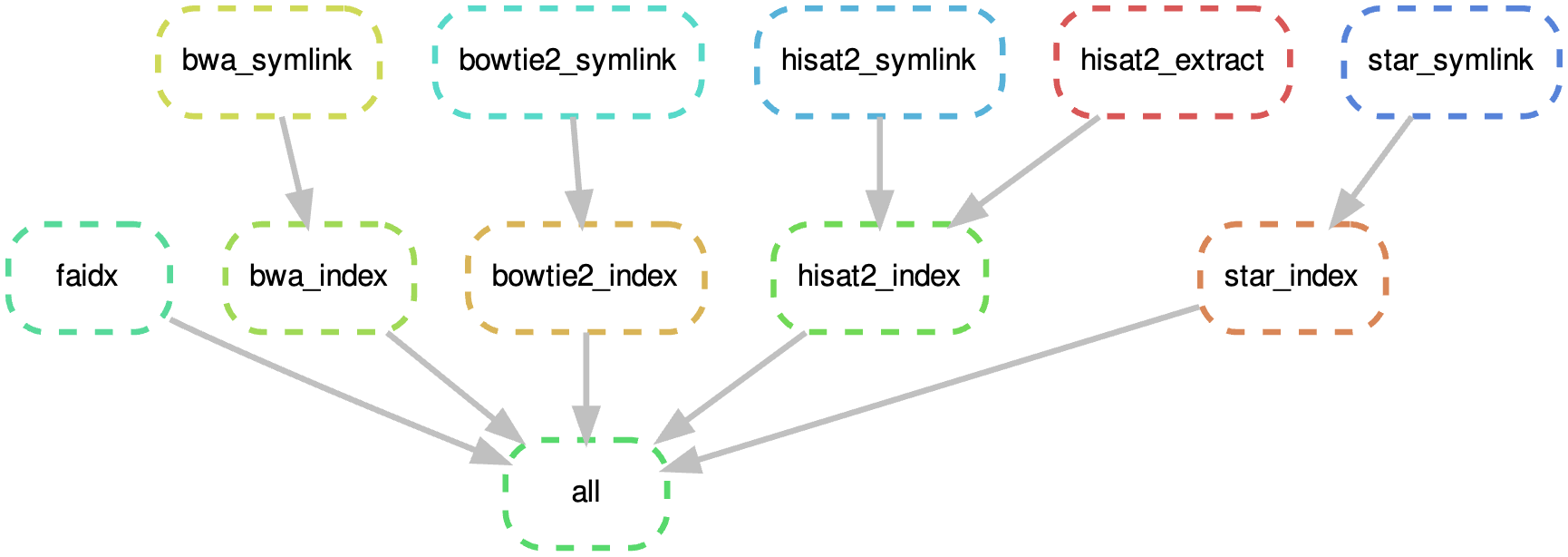
# Snakefile
configfile: "config.yaml"
import os
# 提取参考文件路径
GENOME_FA = config["ref"]["genome"]
GFF3 = config["ref"]["gff3"]
GTF = config["ref"]["gtf"]
# 获取参考基因组目录和文件名
REF_DIR = os.path.dirname(GENOME_FA)
GENOME_BASENAME = os.path.splitext(os.path.basename(GENOME_FA))[0]
# 定义最终目标,确保所有索引文件都将被创建
rule all:
input:
# Samtools faidx 索引
GENOME_FA + ".fai",
# BWA 索引文件
expand(os.path.join(REF_DIR, "bwa_index", os.path.basename(GENOME_FA) + ".{ext}"), ext=["amb", "ann", "bwt", "pac", "sa"]),
# Bowtie2 索引文件
expand(os.path.join(REF_DIR, "bowtie2_index", GENOME_BASENAME + ".{ext}"), ext=["1.bt2", "2.bt2", "3.bt2", "4.bt2", "rev.1.bt2", "rev.2.bt2"]),
# HISAT2 索引文件
expand(os.path.join(REF_DIR, "hisat2_index", GENOME_BASENAME + ".{ext}"), ext=["1.ht2", "2.ht2", "3.ht2", "4.ht2", "5.ht2", "6.ht2", "7.ht2", "8.ht2"]),
# STAR 索引完成标志文件
os.path.join(REF_DIR, "STAR_index", "star_index.done")
# 规则:创建 Samtools faidx 索引
rule faidx:
input:
genome = GENOME_FA
output:
fai = GENOME_FA + ".fai"
shell:
"samtools faidx {input.genome}"
# 规则:创建 BWA 索引
rule bwa_symlink:
input:
genome = GENOME_FA
output:
symlink = os.path.join(REF_DIR, "bwa_index", os.path.basename(GENOME_FA))
run:
import os
os.makedirs(os.path.dirname(output.symlink), exist_ok=True)
if not os.path.exists(output.symlink):
os.symlink(os.path.abspath(input.genome), output.symlink)
rule bwa_index:
input:
genome = GENOME_FA,
symlink = rules.bwa_symlink.output.symlink
output:
expand(os.path.join(REF_DIR, "bwa_index", os.path.basename(GENOME_FA) + ".{ext}"), ext=["amb", "ann", "bwt", "pac", "sa"])
params:
index_prefix = os.path.join(REF_DIR, "bwa_index", os.path.basename(GENOME_FA))
shell:
"""
bwa index -p {params.index_prefix} {input.genome}
"""
# 规则:创建 Bowtie2 索引
rule bowtie2_symlink:
input:
genome = GENOME_FA
output:
symlink = os.path.join(REF_DIR, "bowtie2_index", os.path.basename(GENOME_FA))
run:
import os
os.makedirs(os.path.dirname(output.symlink), exist_ok=True)
if not os.path.exists(output.symlink):
os.symlink(os.path.abspath(input.genome), output.symlink)
rule bowtie2_index:
input:
genome = GENOME_FA,
symlink = rules.bowtie2_symlink.output.symlink
output:
expand(os.path.join(REF_DIR, "bowtie2_index", GENOME_BASENAME + ".{ext}"), ext=["1.bt2", "2.bt2", "3.bt2", "4.bt2", "rev.1.bt2", "rev.2.bt2"])
params:
index_prefix = os.path.join(REF_DIR, "bowtie2_index", GENOME_BASENAME)
shell:
"""
bowtie2-build {input.genome} {params.index_prefix}
"""
# 规则:创建 HISAT2 索引
rule hisat2_symlink:
input:
genome = GENOME_FA
output:
symlink = os.path.join(REF_DIR, "hisat2_index", os.path.basename(GENOME_FA))
run:
import os
os.makedirs(os.path.dirname(output.symlink), exist_ok=True)
if not os.path.exists(output.symlink):
os.symlink(os.path.abspath(input.genome), output.symlink)
rule hisat2_extract:
input:
gtf = GTF
output:
splice_sites = temp(os.path.join(REF_DIR, "hisat2_index", "splicesites.tsv")),
exons = temp(os.path.join(REF_DIR, "hisat2_index", "exons.tsv"))
shell:
"""
hisat2_extract_splice_sites.py {input.gtf} > {output.splice_sites}
hisat2_extract_exons.py {input.gtf} > {output.exons}
"""
rule hisat2_index:
input:
genome = GENOME_FA,
symlink = rules.hisat2_symlink.output.symlink,
splice_sites = rules.hisat2_extract.output.splice_sites,
exons = rules.hisat2_extract.output.exons
output:
expand(os.path.join(REF_DIR, "hisat2_index", GENOME_BASENAME + ".{ext}"), ext=["1.ht2", "2.ht2", "3.ht2", "4.ht2", "5.ht2", "6.ht2", "7.ht2", "8.ht2"])
params:
index_prefix = os.path.join(REF_DIR, "hisat2_index", GENOME_BASENAME)
shell:
"""
hisat2-build --ss {input.splice_sites} --exon {input.exons} {input.genome} {params.index_prefix}
"""
# 规则:创建 STAR 索引
rule star_symlink:
input:
genome = GENOME_FA
output:
symlink = os.path.join(REF_DIR, "STAR_index", os.path.basename(GENOME_FA))
run:
import os
os.makedirs(os.path.dirname(output.symlink), exist_ok=True)
if not os.path.exists(output.symlink):
os.symlink(os.path.abspath(input.genome), output.symlink)
rule star_index:
input:
genome = GENOME_FA,
symlink = rules.star_symlink.output.symlink,
gtf = GTF
output:
marker = os.path.join(REF_DIR, "STAR_index", "star_index.done")
params:
genome_dir = os.path.join(REF_DIR, "STAR_index")
threads: 2
shell:
"""
mkdir -p {params.genome_dir}
STAR --runThreadN {threads} --runMode genomeGenerate --genomeDir {params.genome_dir} \
--genomeFastaFiles {input.genome} --sjdbGTFfile {input.gtf}
touch {output.marker}
"""